
De patofysiologi av depresjon det er basert på forskjeller i hjernestrukturer som størrelsen på amygdala, hippocampus eller prefrontal cortex. Likeledes er det funnet endringer i nevronstørrelse, glia tetthet og metabolisme. Rollen til monoaminer eller andre nevrotransmittere er også dokumentert, og forskjellige teorier har også blitt tilbudt om deres tilblivelse eller forklaring..
Depresjon skyldes ikke utelukkende biologiske eller psykologiske faktorer, men snarere på grunn av den komplekse interaksjonen mellom mange sosiale, psykologiske eller biologiske faktorer.

Når vi ser etter den beste behandlingen for å håndtere depresjon, og tar i betraktning at farmakoterapi (og de forskjellige antidepressiva) også har reagert ugunstig i mange aspekter, har vi søkt hva som er prosessene som er involvert i denne sykdommen..
Artikkelindeks
Tendensen til å utvikle en depressiv lidelse ser ut til å skyldes, på en eller annen måte, arv. Denne informasjonen kommer til oss gjennom familiestudier, slik at en person med en nær slektning med en affektiv lidelse er 10 mer sannsynlig å lide av den enn en annen person som ikke har en berørt pårørende..
Disse dataene indikerer at depressive lidelser har en arvelig tendens. I tillegg kan dette også observeres gjennom studier av monozygotiske tvillinger, som viser at det er større samsvar mellom disse i depresjon enn hos dizygotiske tvillinger..
På samme måte indikerer adopsjons- og depresjonsstudier at det er en høyere forekomst av depresjon hos biologiske foreldre enn hos adoptivforeldre.
I forhold til gener involvert i depresjon, indikerer forskning at det er flere gener involvert, og observerer kobling mellom gener som er lokalisert på kromosomer 2, 10, 11, 17, 18, samt polymorfier av gener som for eksempel serotonintransportøren når det gjelder opprinnelsen til depresjon.
Åpenbart, hvis vi refererer til en sykdom med flere symptomer og hvor variasjonen er stor, er det logisk å tenke at genene som er involvert er også flere..
Flere neuroimaging studier har blitt utført med depressive pasienter som har vist at de presenterer endringer i forskjellige hjernestrukturer. Blant dem fremhever vi endringene i amygdala, i hippocampus og i prefrontal cortex, både dorso-lateral og ventral.
Når det gjelder hippocampus, har noen studier funnet redusert hvit substans og har vist at det er en asymmetri mellom halvkulene, samt mindre volum i begge hippocampus hos pasienter med depresjon.
På det anatomiske nivået er generelt sett at grå materie er redusert i de orbitale og midterste prefrontale cortexområdene, i ventral striatum, i hippocampus og en forlengelse av laterale og tredje ventrikler, noe som innebærer et nevronaltap.
I andre studier, når pasientene var døde, er det funnet et redusert volum av cortex og gliaceller.
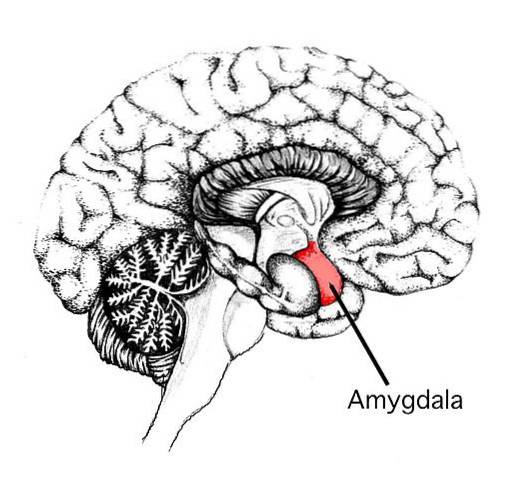
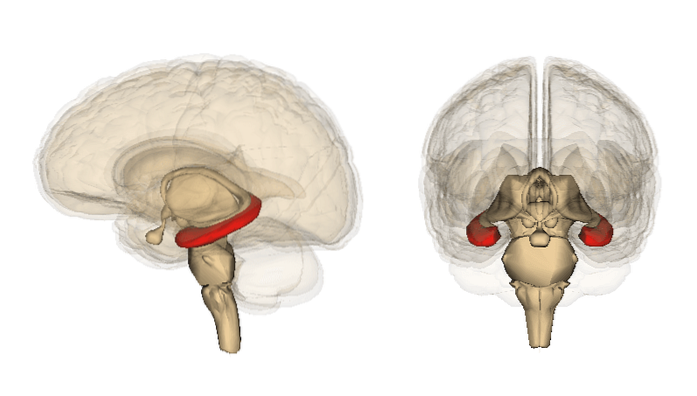
I forhold til amygdala viser studiene variable resultater. Selv om det ikke har vært noen forskjeller når det gjelder volumet på amygdalaen, gjorde noen av dens egenskaper det..
For eksempel forklarte medisinen forskjeller i amygdalavolumet, slik at jo flere personer med medisiner det var i studien, jo større volum av amygdalaen til pasientene med depresjon sammenlignet med kontrollen.
Denne typen resultater kan bidra og forsterke ideen om at depresjon er assosiert med en reduksjon i volumet av amygdala.
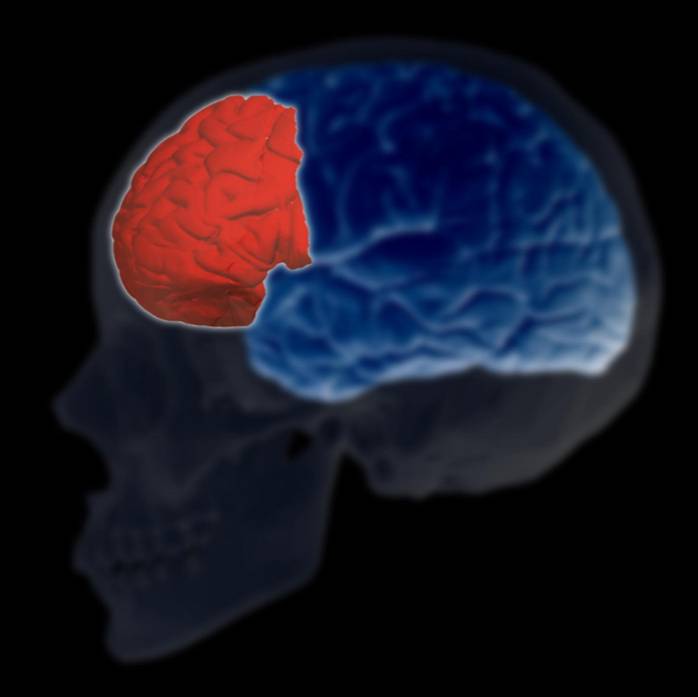
Når det gjelder prefrontal cortex, har flere studier også funnet at pasienter med depresjon hadde mindre volum sammenlignet med kontrollen i rectus gyrus og ikke i andre forskjellige regioner.
Når det gjelder hjerneaktivitet, har nevroavbildningsstudier også vist abnormiteter funnet i blodstrøm og glukosemetabolisme hos deprimerte personer.
Dermed har det blitt antydet at økt metabolisme i amygdala var relatert til en større alvorlighetsgrad av depresjon, mens når metabolsk aktivitet i ventromedial prefrontal cortex ble redusert, er de for reaktive mot indusert tristhet, men hyporeaktive mot depresjon..
I andre studier ble det vist at det var en sammenheng mellom alvorlighetsgraden av depresjon og økt glukosemetabolisme også i andre regioner som det limbiske systemet, ventromedial prefrontal cortex, temporal, thalamus, ventrale områder av basalganglier eller den underordnede parietal cortex.
Tap av motivasjon ved depresjon var også negativt relatert til visse områder, med dorsolateral prefrontal cortex, dorsal parietal cortex, eller dorsotemporal association cortex..
Det var også et forhold i søvn, slik at endringene ble korrelert med større aktivitet i noen kortikale og subkortikale områder.
Det er noen kretsløp som er relatert til depresjon, blant annet kan vi fremheve appetitten og vektøkningen som oppstår hos noen pasienter med depresjon.
Depressivt humør, det viktigste symptomet på depresjon, er relatert til endringer som forekommer i amygdala, i ventromedial prefrontal cortex og i den fremre cingulære gyrus, som involverer både serotonin, dopamin og norepinefrin.
Mangel på energi som også kjennetegner pasienter med depresjon er på sin side relatert til dopamin og noradrenalin og adresserer problemene som finnes i den diffuse prefrontale cortex.
Det er også søvnforstyrrelser relatert til dysfunksjoner i hypothalamus, thalamus, basal forhjernen og hvor noradrenalin, serotonin og dopamin er involvert..
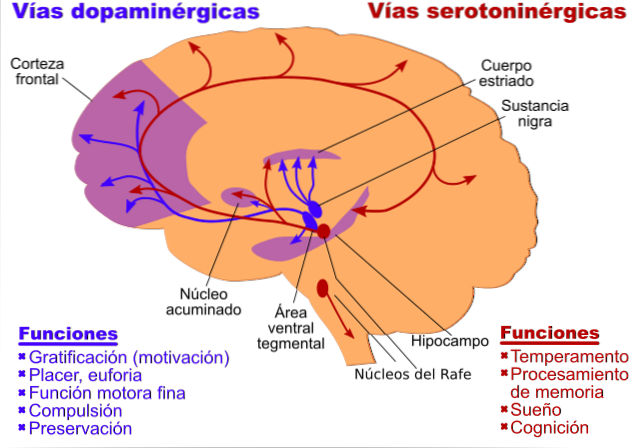
For sin del fant vi at apati er relatert til en dysfunksjon i dorsolaterl prefrontal cortex, nucleus accumbens, og noradrenalin og dopamin er viktige nevrotransmittere..
De psykomotoriske symptomene vi finner i depresjon er assosiert med endringer i striatum, lillehjernen og prefrontal cortex, assosiert med de tre monoaminer.
For sin del er utøvende problemer knyttet til dopamin og noradrenalin og er assosiert med den dorsolaterale prefrontale cortex.
Det er forskjellige teorier eller hypoteser som er samlet rundt depresjonens opprinnelse.
En av dem, den første, oppstår rundt ideen eller hypotesen om at et underskudd av monoaminerge nevrotransmittere, som noradrenalin, dopamin eller serotonin, ville være årsaken til depresjon. Dette er den monoaminergiske hypotesen om depresjon.
Denne hypotesen er basert på forskjellige bevis. En av dem er for eksempel det faktum at reserpin (et medikament for hypertensjon) forårsaket depresjon; det virker ved å hemme lagringen av monoaminer og ved å virke antagonistisk mot monominer. Dermed antydes det at det kan føre til depresjon.
I motsatt tilfelle finner vi medisinene som forbedrer disse nevrotransmitterne og som forbedrer symptomene på depresjon, og fungerer som agonister..
Det skal også bemerkes at det er data som ikke støttet denne hypotesen, og det endelige beviset for at denne hypotesen er det som kalles terapeutisk latens, noe som forklarer den forsinkede forbedringen som oppstår i symptomene på depresjon etter administrering av legemidlet. som indikerer at det må være noen mellomprosess som tar seg av nevnte forbedring.
Det foreslås at det kan være noen annen mekanisme i hjernen som ikke bare tilsvarer monoaminer og som er ansvarlig for depresjon.
En mulig forklarende mekanisme er reseptorene, slik at det ved depresjon kan være en endring av dem, en oppregulering som skyldes det faktum at det er et underskudd av nevrotransmitteren. Ikke produsert nok, over tid er det en økning i antall og følsomhet av reseptorer.
Bevis for denne hypotesen er også funnet, for eksempel studier av selvmordspersonell som gjør at mortem gjør det mulig å finne denne økningen i reseptorer i frontal cortex.
Andre bevis ville være det samme faktum at antidepressiva som tas produserer desensibilisering i reseptorene.
Nyere forskning antyder at det kan være på grunn av en unormalitet i genuttrykket til reseptorene (på grunn av underskudd eller funksjonsfeil).
Andre linjer antyder snarere at det kan være på grunn av en emosjonell dysfunksjon av mekanismer som forandringer i genet for den nevrotrofiske faktoren avledet fra hjernen som støtter levedyktigheten til nevroner.

Ingen har kommentert denne artikkelen ennå.