De Duchenne muskeldystrofi (DMD) er en nevromuskulær sykdom, preget av tilstedeværelsen av betydelig muskelsvakhet, og en generalisert og progressiv utvikling (Verdens helseorganisasjon, 2012).
Det er den vanligste typen muskeldystrofi hos mennesker (López-Hernández, 2009) og rammer 1 av 3500 barn i verden (Duchenne Parent Project, 2012). For det meste rammer sykdommen menn i de tidlige stadiene av livet (Verdens helseorganisasjon, 2012).
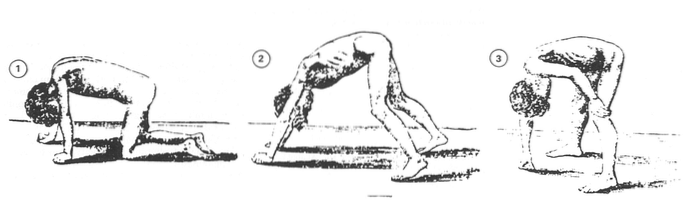
Det finnes forskjellige typer muskeldystrofi. Symptomer begynner vanligvis i barndommen. Svakhet og tap av muskelmasse forårsaker alvorlige vanskeligheter med å tilegne seg eller opprettholde evnen til å gå, puste og / eller svelge (Mayo Clinic, 2013).
Nevromuskulære effekter gir en kronisk prognose. I de fleste tilfeller dør mennesker med Duchenne muskeldystrofi i ung voksen alder på grunn av utvikling av sekundære patologier som hjertesvikt eller kardiomyopatier (Verdens helseorganisasjon, 2012).
Artikkelindeks
Duchenne muskeldystrofi er en sykdom som påvirker individet gjennom progressiv muskelsvakhet og degenerasjon (Muscular Dystrophy Association, 2016).
På grunn av en genetisk mutasjon vil fraværet av et spesifikt protein hos mennesker med Duchenne muskeldystrofi føre til tap av muskelfunksjonalitet..
Vanligvis vises symptomene i underekstremitetene og spres til resten av områdene.
Verdens helseorganisasjon (2012) indikerer at forekomsten av Duchenne muskeldystrofi er anslått til omtrent 1 tilfelle per 3300 innbyggere.
Spesielt viser noen undersøkelser at denne sykdommen rammer 1 av 3500 mannlige barn født i live (López-Hernández, 2009).
I USAs tilfelle er det ikke kjent med sikkerhet hvor mange mennesker i alle aldersgrupper som lider av denne sykdommen. Noen undersøkelser har anslått at en av 5600-7 770 mannlige voksne i alderen 5 til 24 år har en diagnose av Duchenne eller Becker muskeldystrofi (Centers for Disease Control and Prevention, 2015).
Den mest karakteristiske for forstyrrelsene som tilhører gruppen av muskeldystrofi er muskelsvakhet; Avhengig av type kan det imidlertid oppstå spesifikke symptomer som vil variere avhengig av alder på begynnelsen og de berørte muskelgruppene (Mayo Clinic, 2013).
Normalt er utviklingen av Duchnne muskeldystrofi ganske forutsigbar. Foreldre kan observere noen ganske signifikante tegn, for eksempel vanskeligheter eller manglende evne til å lære å gå eller unormal økning i leggmuskulaturen (pseudohypertrofi) (Duchenne foreldreprosjekt, 2012).
Noen av de mest karakteristiske symptomene og tegnene på Duchenne muskeldystrofi som dukker opp tidlig i et barns liv er (Mayo Clinic, 2013):
Tilsvarende fremhever foreningen Duchenne Parent Projet (2012) de vanligste symptomene og kliniske manifestasjonene:
Alle muskelsymptomer begynner med svakhet i bekkenbeltets muskler, kalver og forskjellige gangforstyrrelser som er signifikante før 5 år (López-Hernández, 2009).
I førskolen kan barn med Duchennes muskeldystrofi falle ofte eller ha vanskeligheter med å gå, klatre trinn og / eller løpe (Duchenne Parent Project, 2012).
Etter hvert som sykdommen utvikler seg, i skolealder, er det ganske sannsynlig at barn bare bruker spissene på føttene til å gå. Vi vil være i stand til å observere en rullende og usikker marsj som kan forårsake mange fall. De bruker vanligvis noen strategier for å opprettholde balansen, for eksempel å skyve skuldrene tilbake eller holde på sin egen kropp (Duchenne Parent Project, 2012).
Rundt 9 år klarer de fleste med denne sykdommen ikke å gå, på grunn av dette begynner de å utvikle mange muskuloskeletale misdannelser - skoliose, kontrakturer osv. - (López-Hernández, 2009).
I ungdomsfasen vil de presentere betydelige vanskeligheter med å effektivt utføre aktiviteter knyttet til bruk av øvre ekstremiteter, ben eller koffert. På dette stadiet vil de kreve mekanisk støtte og hjelp (Duchenne Parent Project, 2012).
Muskeldegenerasjon og svakhet fortsetter å gå frem til de når musklene som er ansvarlige for luftveis- og hjertefunksjon (López-Hernández, 2009). På grunn av alt dette er pasientens overlevelse alvorlig kompromittert og forårsaker død i de fleste tilfeller..
Det er identifisert forskjellige gener som er involvert i produksjonen av proteiner som er ansvarlige for å beskytte muskelfibre mot mulig skade og skade (Mayo Clinic, 2013).
Spesielt oppstår hver type muskeldystrofi som en konsekvens av en bestemt genetisk mutasjon. Noen av disse mutasjonene arves; i de fleste tilfeller forekommer de imidlertid spontant under graviditet (Mayo Clinic, 2013).
Når det gjelder Duchenne muskeldystrofi, identifiserte forskerne et spesifikt gen lokalisert på X-kromosomet som kunne presentere mutasjonen som var ansvarlig for denne patologien (Muscular Dystrophy Association, 2016).
I 1987 ble proteinet assosiert med dette genet således identifisert., dystrofin. Dermed innebærer mangel eller fravær av dette proteinet at musklene er skjøre og lett skadet (Muscular Dystrophy Association, 2016).
I tillegg er det identifisert et recessivt arvemønster knyttet til X-kromosomet, med bæreren som mor (Muscular Dystrophy Association, 2016). På grunn av dette faktum er denne typen sykdom hyppigere hos menn enn hos kvinner..
Menn har en XY-kromosomsammensetning, mens kvinner er XX. Derfor, hvis et X-kromosom har en mutasjon i DMD-genet, vil det lide av Duchennes muskeldystrofi på grunn av fravær av dystrofinproduksjon (National Human Genome Research Institute, 2013).
Imidlertid, når det gjelder kvinner som har to X-kromosomer og derfor to kopier av DMD-genet, hvis en av disse endres, vil den andre kunne fortsette å produsere dystrofin og derfor opprettholde muskelneurbeskyttelse (National Human Genome Research Institute, 2013 ).
I denne typen patologier kan forskjellige inngrep utføres for å bestemme diagnosen (National Human Genome Research Institute, 2013).
Den kliniske diagnosen kan allerede stilles når et barn begynner å utvikle progressiv muskelsvakhet. Allerede ved 5 års alder er det åpenbare symptomer. Hvis det ikke utføres en tidlig intervensjon, vil barn ha funksjonell avhengighet før fylte 13 år (National Human Genome Research Institute, 2013).
Bortsett fra observasjon og klinisk undersøkelse, kan noen av følgende teknikker brukes til å identifisere tilstedeværelsen av Duchenne muskeldystrofi (Mayo Clinic, 2013):
For tiden er det ikke identifisert en kur mot Duchenne muskeldystrofi (Duchenne Parent Project, 2012).
Til tross for dette brukes forskjellige behandlinger som har vist seg å være effektive både for å redusere symptomer og for å forbedre livskvaliteten til mennesker som lider av denne typen patologi (Duchenne Parent Project, 2012).
På grunn av den kliniske progresjonen og de mange symptomene vil denne sykdommen kreve en tverrfaglig og omfattende intervensjon utført av et bredt utvalg av spesialister: barnelege, fysioterapeut, nevrolog, nevropsykolog, ergoterapeut, logoped, ernæringsfysiolog, endokrinolog, genetiker, kardiolog, pulmonolog, ortoped, rehabilitator og kirurg, blant andre (Duchenne Parent Project, 2012).
I mange tilfeller kan spesialister anbefale farmakologiske inngrep (Mayo Clinic, 2013):
Ikke bare er medisiner nyttige for intervensjon i Duchenne muskeldystrofi, det er både terapeutiske inngrep og pleiemetoder som kan forbedre livskvaliteten til disse menneskene (Mayo Clinic, 2013).
Noen fordelaktige inngrep er (Duchenne Parent Project, 2012):
Inntil for relativt få år siden overlevde ikke mennesker med Duchenne muskeldystrofi mye lenger etter å ha nådd ungdomsårene (Muscular Dystrophy Association, 2016).
De store fremskrittene innen medisinsk, teknisk og genetisk forskning har klart å redusere sykdommens progresjon og å gi en betydelig økning i livskvaliteten til personer som lider av den (Muscular Dystrophy Association, 2016). På denne måten er hjerte- og åndedrettspleie viktig for bevaring av vitale funksjoner (Muscular Distrophy Association, 2016).
I mange tilfeller er de i stand til å nå stadiene etter ungdomsårene. Flere og flere tilfeller av Duchenne muskeldystrofi blir beskrevet hos voksne i 30-årene, inkludert mennesker som overlever til 40- og 50-årene (Muscular Dystrophy Associatin, 2016).
For tiden er kliniske studier og forskning orientert mot utvikling av genterapier som modifiserer mutasjoner og mangler i dystrofinproduksjonen (Muscular Dystrophy Association, 2016).
Noen av de mest etterforskede mekanismene er (López-Hernández, 2009):
Duchennes muskeldystrofi er en alvorlig invalidiserende sykdom hos både barn og unge voksne med en ødeleggende prognose..
Til tross for at klinisk og eksperimentell forskning har gjort viktige fremskritt i behandlingen av symptomer, er det fortsatt ingen kur for denne typen patologi..
En grundig forståelse av det biologiske og genetiske grunnlaget er viktig for å finne en kurativ behandling for Duchenne muskeldystrofi..



Ingen har kommentert denne artikkelen ennå.