De prioner de er proteiner uten genom eller nukleinsyrer som fungerer som smittsomme midler. Uttrykket "prion" betyr proteinholdig smittsom partikkel (fra engelsk Proteinaceous Infectious Particles), og ble laget av nevrologen og Nobelprisvinneren, Stanley B. Prusiner.
I 1982 identifiserte Prusiner og hans kolleger en smittsom proteinpartikkel, mens de studerte årsakene til Creutzfeldt-Jakobs sykdommer (hos mennesker) og bovin spongiform encefalopati..
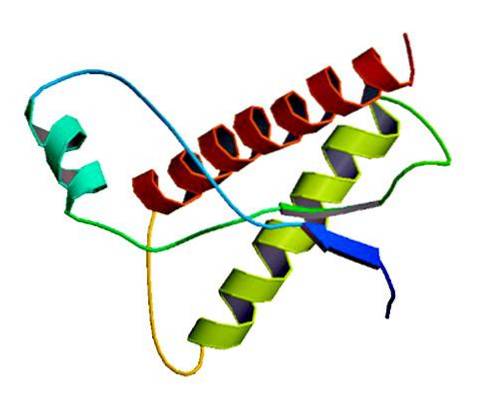
Disse sjeldne smittsomme stoffene finnes i membranen til normale celler, bare som feilfoldede proteiner og / eller med en unormal tredimensjonal struktur. Disse proteinene er ansvarlige for flere degenerative sykdommer og svært høy dødelighet som påvirker nevrale vev og hjernens struktur..
De kalles også prionsykdommer. Blant de viktigste som påvirker mennesker er kuru, Gerstmann-Sträussler-Scheinker sykdom, Creutzfeldt-Jakob syndrom og dødelig familiær søvnløshet..
Artikkelindeks
Prioner er proteinstrukturer som er tilstede i cellemembraner. Disse proteinene har endret form eller konformasjon [PrP (Sc)].
Når det gjelder multiplikasjon, oppnås det gjennom konvertering av former, som i tilfelle av skrapesykdom. I denne sykdommen rekrutterer prioner PrP (C) (prionproteiner med uendret konformasjon) for å stimulere konvertering til PrP (Sc) isoformen..
Dette genererer en kjedereaksjon som sprer det smittsomme materialet og derfor tillater vanning av sykdommen. Det er fortsatt ukjent hvordan denne konverteringsprosessen skjer.
Disse uvanlige proteinene som kan spre seg, har ikke nukleinsyrer. Bevis på dette er at de er motstandsdyktige mot røntgen og ultrafiolett stråling. Disse midlene bryter lett ned nukleinsyrer.
Prion-proteiner, hvorav prioner (PrP) er sammensatt, finnes i hele kroppen, ikke bare hos mennesker, men hos andre sunne virveldyr. Disse proteinene er generelt motstandsdyktige mot proteaser (enzymer som katalyserer proteiner).
Svært lite er kjent om nytten av prionproteinene PrP (C), den normale formen for det ikke-smittsomme proteinet i menneskekroppen..
Noen forskere har imidlertid lykkes med å vise at disse musene aktiverer myelinreparasjon i celler i det perifere nervesystemet hos mus. Fraværet av disse har også vist seg å forårsake demyelinisering av slike nerveceller..
Kunnskapen som finnes om strukturen til prioner ligger hovedsakelig i undersøkelsene som er utført i bakterien Escherichia coli.
Studier har vist at kjedepolypeptidene PrP (C) (normal) og PrP (Sc) (smittsom) er identiske med hensyn til aminosyresammensetning, men er forskjellige i 3D-konformasjon og folding..
Disse ikke-smittsomme prionene har 209 aminosyrer hos mennesker. De har en disulfidbinding. Dens struktur er alfa-spiralformet, noe som betyr at den har spiralformede aminosyrer (alpha helices) og få flate tråder av aminosyrer (beta-ark)..
Dette proteinet kan ikke skilles fra ved sentrifugering, noe som betyr at det ikke er sedimenterbart. Det fordøyes lett av bredspektret serinprotease kalt proteinase K.
Det er et smittsomt protein som transformerer PrP (C) til smittsomme PrP (Sc) isoformer og med en unormal konfigurasjon eller form.
Svært lite er kjent om 3D-strukturen, men det er kjent at den har få spiralformede former og flere flate tråder eller beta-ark. Skiftet til isoformen er det som er kjent som den viktigste hendelsen av prionsykdommer.
Cellulære prionproteiner [Prp (C)] er lokalisert på celleoverflaten til et bredt utvalg av organer og vev. Svært lite er kjent om de fysiologiske funksjonene til prioner i kroppen. Allikevel indikerer eksperimenter gjort på mus mulige funksjoner, for eksempel:
PrP (C) har vist seg å virke med glutamatreseptorer (ionotrope og metabotrope). PrP (C) deltar som en reseptor for synaptotoksiske oligomerer av celleoverflate-peptidet Aβ.
Hos mus av Murinae-familien har det blitt oppdaget at prionproteinene PrP (C) uttrykkes i løpet av få dager etter implantering, i embryonal utvikling.
Dette indikerer at de spiller en rolle under utviklingen av disse små pattedyrene. Roll som ifølge forskerne er relatert til regulering av neuritogenese (produksjon av aksoner og dendritter av nevroner).
De virker også på aksonal vekst. Disse prionproteinene er til og med involvert i utviklingen av lillehjernen. På grunn av dette antas det at fraværet av disse PrP (C) -prionene medfører en forsinkelse i motorisk utvikling av gnagere..
I studier om overekspresjon av PrP (C) ved genorientering, ble det funnet at fraværet av disse prionene forårsaker problemer med blodtilførselen til noen deler av hjernen (akutt cerebral iskemi).
Dette betyr at prionproteiner fungerer som nevrobeskyttere. I tillegg har det blitt vist at overuttrykk av PrP (C) kan redusere eller forbedre skader forårsaket av iskemi..
Den fysiologiske rollen til Prp (C) i vedlikehold av perifert myelin ble nylig oppdaget.
I løpet av en laboratoriestudie ble det oppdaget at i fravær av prionproteinet utviklet laboratoriemus mangler i nervene som bærer informasjon fra hjernen og ryggmargen, i det som kalles perifer nevropati..
Det er noen proteiner som ligner på prioner, og disse ligger i andre deler av kroppen enn hjernen.
Funksjonene til slike proteiner er å initiere, regulere og / eller kontrollere celledød når organismen blir angrepet (for eksempel av vironer), og dermed forhindre spredning av patogenet..
Denne særegne funksjonen til disse proteinene får forskere til å tenke på muligheten for ikke-smittsomme prioner i kampen mot patogener..
En studie utført ved Stowers Institute, i Missouri, USA viste at PrP-prioner kan ha en rolle i å opprettholde langtidsminnet.
Studien avslørte at visse prionproteiner kan kontrolleres for å opprettholde de fysiologiske funksjonene til langtidshukommelsen..
En undersøkelse av prionproteiner som uttrykkes i stamceller i blodvev, avslørte at alle disse stamcellene (hematopoietiske) uttrykker prionproteiner i cellemembranen. For hva det antas at de deltar i den komplekse og veldig viktige prosessen med cellefornyelse.
Patologier av prion opprinnelse er anerkjent som progressive degenerative hjernesykdommer. De kan angripe storfe, hjort, karibou, sauer og til og med mennesker.
Disse sykdommene er forårsaket av en endring i strukturen til PrP (C) proteiner, og hvis spesifikke funksjoner fremdeles er usikre i dag. Prionpatologier kan oppstå uten kjent årsak. De kan ha arvelig genetisk opprinnelse og kan også overføres på en smittsom-smittsom måte.
Prions forårsaker familiære, sporadiske og smittsomme sykdommer. Familiale prionsykdommer er de som er arvelige. Sporadiske patologier er de vanligste og forekommer uten kjente årsaker..
Smittsomme sykdommer regnes som sjeldne, de overføres av person til person, dyr til dyr, person til dyr og omvendt. Årsakene er flere og spenner fra forbruk av forurenset kjøtt, kannibalisme, transfusjoner, til manipulering av forurenset kirurgisk utstyr.
De vanligste prionsykdommene er:
Ansett som den vanligste prionsykdommen blant mennesker, er det en kosmopolitisk sykdom, det vil si at den har en verdensomspennende distribusjon. Det kan være arvelig (familiær), sporadisk eller smittsom.
Pasienter som har symptomer som demens, rykk eller plutselige ufrivillige bevegelser og mangler i sentralnervesystemet.
Avhengig av sykdomsbehandlingen og formen, kan døden oppstå mellom 4 måneder og 2 år etter anskaffelsen av sykdommen. Diagnose er vanskelig å stille, det gjøres vanligvis post morten, under obduksjon.
Det er en sykdom forårsaket av prioner i en arvelig eller autosomal dominerende smittsom hjerneprosess. Sykdommen manifesterer seg hos mennesker i alderen 40 til 60 år.
Disse menneskene viser problemer med å artikulere ord (dysartri), rykk eller plutselige ufrivillige bevegelser, aggressivitet er ofte.
De presenterer med cerebellar degenerasjon ledsaget av en ustabil gangart. Det er også mulig å observere hyporefleksi, døvhet, blikk lammelse, demens, blant andre symptomer. Forventet levetid er omtrent 5 år eller litt lenger.
Det er en veldig sjelden sykdom, til det punktet at forekomstområdet er 2 til 3 tilfeller per 100 millioner innbyggere. Patologien ligner på Gerstmann-Sträussler-Scheinkers sykdom.
Proteinens kliniske manifestasjoner indikerer lav motstand mot proteaser, noen er mer og andre mindre følsomme for disse enzymene.
Symptomene pasienter presenterer er: problemer med tale og kognitiv svikt, tap av nevroner i området der hjernen styrer bevegelser og utfører muskelkoordinering.
Sykdommen er hyppig hos eldre pasienter (70 år), og den estimerte levetiden når den er smittet er omtrent 20 måneder.
Det er en arvelig eller familiær sykdom, den kan også forekomme sporadisk. Det er kjent at sykdommen skyldes en arvelig eller autosomal dominant mutasjon.
Pasienter presenterer symptomer som kumulative problemer med å sove og opprettholde søvn, demens, kognitiv svikt, til og med problemer med høyt blodtrykk, takykardi, hyperhidrose og andre..
Alderen det påvirker er ganske bred, og varierer mellom 23 og 73 år, men gjennomsnittsalderen er 40 år. Levetiden en gang smittet er litt over 6 år.
Denne prionsykdommen har bare blitt oppdaget hos innbyggerne i Papua Ny-Guinea. Det er en sykdom relatert til kannibalisme og den kulturelle tradisjonen for riten om å sørge de døde, der disse menneskene spiser hjerne eller menneskekjøtt..
Mennesker som bærer sykdommen har vanligvis ukontrollerbare og ufrivillige bevegelser i forskjellige deler av kroppen.
De presenterer skjelving, tap av kontroll over bevegelser og tap av muskelkoordinasjon. Forventet levetid hos smittede mennesker er to år.
Blant patologiene produsert av prioner hos dyr er bovin spongiform encefalopati. Denne sykdommen forårsaket kaos i Europa, i folkehelsen, hos dyr og i økonomien i de berørte landene.
Andre sykdommer hos dyr inkluderer skrapesyke, overførbar minkeencefalopati, kronisk bortkastet sykdom (hos hjort) og feline spongiform encefalopati..
Disse sykdommene, som de som presenteres hos mennesker, mangler effektiv behandling, så forebygging er viktig, spesielt etter infeksjoner hos mennesker som har oppstått som et resultat av forbruk av kjøtt fra infiserte kyr..
Til dags dato er det ingen kjent kur mot prionsykdommer. Behandlingen er symptomatisk. Pasienter anbefales å planlegge lindrende behandling, og det anbefales genetisk testing og rådgivning for familiemedlemmer.
Et bredt utvalg av medisiner er testet hos pasienter med prionsykdommer, som antivirale midler, antitumorer, medisiner mot sykdommer som Parkinsons, behandlinger for immunsuppresjon, antibiotika, antimykotika, til og med antidepressiva..
Imidlertid er det foreløpig ingen bevis som indikerer at noen av disse reduserer symptomene eller forbedrer pasientens overlevelse..
Prioner er motstandsdyktige mot en rekke fysiske og kjemiske endringer. Imidlertid brukes forskjellige teknikker for å unngå forurensning av pasienter med forurensede kirurgiske instrumenter..
Blant de mest brukte teknikkene er å sterilisere utstyret i en autoklav ved 132 ° C i en time og deretter dyppe instrumentene i natriumhydroksid i minst en time til..
På den annen side har verdens helseorganisasjon (WHO) utviklet tiltak for å forhindre spredning av prionsykdommer. Denne organisasjonen fastsetter normer for håndtering av forbudte eller potensielt risikable vev som: øyne, hjerne, tarm, mandler og ryggmarg.



Ingen har kommentert denne artikkelen ennå.