De formell tiltale (CF) er et som er tilordnet et atom av et molekyl eller ion, som gjør det mulig å forklare dets strukturer og kjemiske egenskaper basert på det. Dette konseptet innebærer vurdering av maksimal karakter av kovalens i bindingen A-B; det vil si at elektronparet deles likt mellom A og B.
For å forstå det ovennevnte viser det nedre bildet to sammenkoblede atomer: ett betegnet med bokstaven A og det andre med bokstaven B. Som det fremgår, dannes det i skjæringspunktet mellom sirklene en binding med paret ":". I dette heteronukleære molekylet, hvis A og B har like elektronegativiteter, forblir paret ":" like langt fra både A og B.
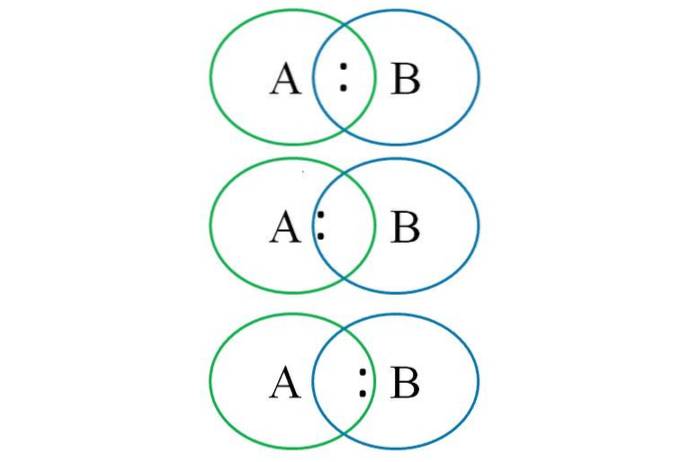
Men siden to forskjellige atomer ikke kan ha identiske egenskaper, tiltrekkes ":" - paret av den som er mer elektronegativ. I dette tilfellet, hvis A er mer elektronegativ enn B, er paret ":" nærmere A enn B. Det motsatte oppstår når B er mer elektronegativ enn A, nå nærmer seg ":" til B.
Så for å tildele de formelle anklagene til både A og B, er det nødvendig å vurdere den første saken (den over bildet). Hvis den rent kovalente bindingen A-B ble brutt, ville det oppstå et homolytisk brudd som genererte de frie radikaler A · og · B.
Artikkelindeks
Elektronene er ikke faste, som i forrige eksempel, men vandrer og går tapt gjennom atomene i molekylet eller ionet. Hvis det er et diatomisk molekyl, er det kjent at paret ":" må deles eller vandre mellom begge atomene; det samme skjer i et molekyl av typen A-B-C, men med større kompleksitet.
Men når man studerer et atom og antar en kovalens på hundre prosent i bindingene, er det lettere å fastslå om det får eller mister elektroner i forbindelsen. For å bestemme denne gevinsten eller tapet, må baseline eller fri tilstand sammenlignes med det elektroniske miljøet ditt..
På denne måten er det mulig å tilordne en positiv ladning (+) hvis atomet mister et elektron, eller en negativ ladning (-) når det tvert imot får et elektron (tegnene må skrives inne i en sirkel).
Så selv om elektronene ikke kan lokaliseres nøyaktig, samsvarer disse formelle ladningene (+) og (-) på strukturene i de fleste tilfeller med de forventede kjemiske egenskapene..
Det vil si at den formelle ladningen til et atom er nært knyttet til molekylgeometrien i omgivelsene og dens reaktivitet i forbindelsen..
Er formelle anklager tildelt vilkårlig? Svaret er nei. For dette må gevinst eller tap av elektroner beregnes forutsatt rent kovalente bindinger, og dette oppnås gjennom følgende formel:
CF = (gruppens nummer på atomet) - (antall bindinger det danner) - (antall ikke delte elektroner)
Hvis atomet har en CF med verdien +1, tildeles den en positiv ladning (+); mens hvis du har en CF med verdien -1, tildeles den en negativ ladning (-).
For å beregne CF korrekt må følgende trinn følges:
- Finn i hvilken gruppe atomet finnes i det periodiske systemet.
- Tell antall obligasjoner den danner med sine naboer: dobbeltbindinger (=) er verdt to og tredobbelte er verdt tre (≡).
- Til slutt teller antall ikke-delte elektroner, som lett kan observeres med Lewis-strukturer.
Gitt det lineære molekylet A-B-C-D, kan de formelle ladningene for hvert atom variere hvis strukturen for eksempel nå er skrevet som: B-C-A-D, C-A-B-D, A-C-D-B, etc. Dette er fordi det er atomer som, ved å dele flere elektroner (danne flere bindinger), får positiv eller negativ CF.
Så hvilken av de tre mulige molekylære strukturene tilsvarer forbindelse ABCD? Svaret er: den som generelt har de laveste CF-verdiene; på samme måte den som tildeler negative ladninger (-) til de mest elektronegative atomer.
Hvis C og D er mer elektronegative enn A og B, får de følgelig formelle positive ladninger (sett fra en mnemonisk regel) ved å dele flere elektroner..
Dermed er den mest stabile strukturen, og den mest energisk favoriserte, C-A-B-D, siden både C og B bare danner en binding. På den annen side er strukturen A-B-C-D og de som har C eller B som danner to bindinger (-C- eller -D-), mer ustabile.
Hvilken av alle strukturene er den mest ustabile? A-C-D-B, fordi ikke bare C og D danner to bindinger, men også deres formelle negative ladninger (-) ligger ved siden av hverandre, noe som ytterligere destabiliserer strukturen.
Boratomet er omgitt av fire fluoratomer. Siden B tilhører gruppe IIIA (13), mangler den ikke delte elektroner og danner fire kovalente bindinger, og dens CF er (3-4-0 = -1). På den annen side, for F, et element i gruppe VIIA (17), er dens CF (7-6-1 = 0).
For å bestemme ladningen til ionet eller molekylet, er det nok å legge til den enkelte CF av atomene som komponerer den: (1 (-1) + 4 (0) = -1).
CF for B har imidlertid ingen reell betydning; det vil si at den høyeste elektrondensiteten ikke ligger på den. I virkeligheten fordeles denne elektrontettheten mot de fire atomene i F, et element som er mye mer elektronegativt enn B.
Berylliumatomet tilhører gruppe IIA (2), danner to bindinger og mangler igjen ikke delte elektroner. Dermed er CF-ene for Be og H:
CFVære= 2-2-0 = 0
CFH= 1-1-0 = 0
BeH belastningto= 1 (0) + 2 (0) = 0
Dens Lewis-struktur kan representeres som: C20: (selv om den har andre resonansstrukturer). Gjenta CF-beregningen, denne gangen for C (av gruppe IVA) og O (for gruppe VIA), har vi:
CFC= 4-3-2 = -1
CFELLER= 6-3-2 = +1
Dette er et eksempel der formelle avgifter ikke samsvarer med elementenes natur. O er mer elektronegativ enn C og bør derfor ikke ha et positivt.
De andre strukturene (C = O og (+)CO(-)), selv om de overholder den sammenhengende tildelingen av ladninger, overholder de ikke oktettregelen (C har mindre enn åtte valenselektroner).
jo flere elektroner N deler, jo mer positiv er CF-en (til og med ammoniumionen, siden den ikke har energitilgjengelighet for å danne fem bindinger).
På samme måte som beregningene for N i ammoniumion, ammoniakk og amidion, har vi da:
CF = 5-4-0 = +1 (NH4+)
CF = 5-3-2 = 0 (NH3)
Og endelig:
CF = 5-2-4 = -1 (NHto-)
Det vil si i NHto- N har fire ikke-delte elektroner, og deler dem alle når den danner NH4+. CF for H er lik 0 og derfor blir beregningen din lagret.



Ingen har kommentert denne artikkelen ennå.