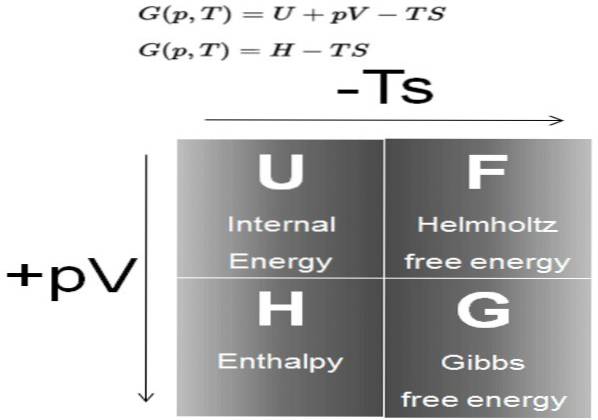
De Gibbs fri energi (ofte kalt G) er et termodynamisk potensial definert som differansen til entalpi H, minus produktet av temperaturen T, av systemets entropi S:
G = H - T S
Gibbs fri energi måles i Joule (i henhold til det internasjonale systemet), i erger (for Cegesimal System of Units), i kalorier eller i elektronvolt (for elektro volt).
I prosesser som oppstår ved konstant trykk og temperatur, er variasjonen av Gibbs fri energi ΔG = ΔH - T ΔS. I slike prosesser representerer (G) energien som er tilgjengelig i systemet som kan konverteres til arbeid.
For eksempel i eksotermiske kjemiske reaksjoner avtar entalpi mens entropi øker. I Gibbs-funksjonen motvirkes disse to faktorene, men bare når Gibbs-energien synker, oppstår reaksjonen spontant..
Så hvis variasjonen av G er negativ, er prosessen spontan. Når Gibbs-funksjonen når sitt minimum, når systemet en stabil likevektstilstand. Oppsummert, i en prosess der trykk og temperatur forblir konstant, kan vi bekrefte:
- Hvis prosessen er spontan, så ΔG < 0
- Når systemet er i likevekt: ΔG = 0
- I en ikke-spontan prosess øker G: ΔG> 0.
Artikkelindeks
Gibbs frie energi (G) beregnes ved å bruke definisjonen gitt i begynnelsen:
G = H - T⋅S
I sin tur er entalpi H et termodynamisk potensial definert som:
H = U + P V
Deretter vil det bli gjort en trinnvis analyse for å kjenne de uavhengige variablene som Gibbs-energien er en funksjon av:
1 - Fra den første loven om termodynamikk har vi at den indre energien U er relatert til entropien S i systemet og dets volum V for reversible prosesser gjennom differensialforholdet:
dU = dQ - dW = TdS - PdV
Fra denne ligningen følger det at den indre energien U er en funksjon av variablene S og V:
U = U (S, V)
2 - Ut fra definisjonen av H og tar differensial, får vi:
dH = dU + d (P V) = dU + VdP + PdV
3- Ved å erstatte uttrykket for dU oppnådd i (1) har vi:
dH = TdS - PdV + VdP + PdV = TdS + VdP
Fra dette konkluderes det at entalpi H avhenger av entropien S og trykket P, det vil si:
H = H (S, P)
4- Nå beregnes den totale differensialen av Gibbs fri energi for å oppnå:
dG = dH -TdS -SdT = TdS + VdP -TdS -SdT
Der dH er erstattet av uttrykket i (3).
5- Til slutt, når vi forenkler, får vi: dG = VdP - SdT, å være klar over at den frie energien G avhenger av trykket og temperaturen T som:
G = G (P, T)
Fra analysen i forrige avsnitt følger det at den indre energien til et system er en funksjon av entropi og volum:
U = U (S, V)
Så differensialet av ELLER vil være:
dU = ∂SU |V dS + ∂VU |S dV = TdS - PdV
Fra dette delvis avledede uttrykket kan de såkalte Maxwell termodynamiske relasjonene utledes. Delderivater gjelder når en funksjon er avhengig av mer enn en variabel og beregnes enkelt ved å bruke teoremet i neste avsnitt.
∂VT |S = -∂SP |V
For å komme til dette forholdet, er Clairaut-Schwarz-setning på delderivater, som sier følgende:
"De blandede derivatene av andre orden med de utskiftede variablene er like, så lenge funksjonene som skal utledes er kontinuerlige og differensierbare".
Basert på det som er vist i punkt 3 i forrige avsnitt:
H = H (S, P) og dH = TdS + VdP
Det kan fås:
∂PT |S = ∂SV |P
Vi fortsetter på en lignende måte med Gibbs fri energi G = G (P, T) og med Helmholtz fri energi F = F (T, V) for å oppnå de to andre termodynamiske forholdene fra Maxwell.

1- Assosiert med den interne energien U: ∂VT |S = -∂SP |V
2- Den som fås fra entalpi H: ∂PT |S = ∂SV |P
3- Relatert til Helmholtz-energien F: ∂TP |V = ∂VS |T
4- Koblet til Gibbs fri energi G: ∂TV |P = -∂PS |T
Beregn variasjonen av Gibbs fri energi for 2 mol ideell gass ved en temperatur på 300K under en isoterm ekspansjon som tar systemet fra et initialvolum på 20 liter til et sluttvolum på 40 liter.
Minner om definisjonen av Gibbs fri energi vi har:
G = H - T S
Da vil en endelig variasjon av F være:
ΔG = ΔH - T ΔS, siden ΔT = 0
I ideelle gasser avhenger entalpi bare av den absolutte temperaturen, men siden det er en isoterm prosess, er ΔH = 0 og ΔG = - T ΔS.
For ideelle gasser er entropiendringen av en isotermisk prosess:
ΔS = nR ln (V.to/ V1)
Det som gjelder tilfellet med denne øvelsen gjenstår:
ΔS = 2 mol x 8,314 J / (K mol) x ln (40L / 20L) = 11,53 J / K
Da kan vi få endringen i Helmholtz energi:
ΔG = - 300K x 11,53 J / K = -3457,70 J.
Med tanke på at Gibbs fri energi er en funksjon av temperatur og trykk G = G (T, P); bestemme variasjonen av G under en prosess der temperaturen ikke endres (isotermisk) for n mol av en monatomisk idealgass.
Som vist ovenfor avhenger endringen i Gibbs energi bare av endringen i temperatur T og volum V, så en uendelig liten variasjon av den blir beregnet i henhold til:
dG = -SdT + VdP
Men hvis det er en prosess der temperaturen er konstant, da dF = + VdP, så en endelig trykkvariasjon AP fører til en endring i Gibbs-energien gitt av:
ΔG = + ∫ VdP = + ∫ (n R T) dP / P = + n R T ln (AP)
Ved hjelp av den ideelle gassligningen:
P V = n R T
Under en isotermisk prosess oppstår det at:
d (P V) = P dV + V dP = 0
Det er:
dP / P = - dV / V.
Så resultatet ovenfor kan skrives som en funksjon av volumvariasjonen AV:
ΔG = + ∫ VdP = + ∫ (n R T) dP / P = - ∫ (n R T) dV / V = -n R T ln (AV)
Tatt i betraktning følgende kjemiske reaksjon:
Nto0 (g) + (3/2) Oto (g) ↔️ 2NOto (g) ved temperatur T = 298 K
Finn variasjonen av Gibbs fri energi, og bruk resultatet oppnådd, om det er en spontan prosess eller ikke.
Her er trinnene:
- Trinn 1: Enthalpies of Reaction
ΔHr = 2 * ΔH (NOto (g)) - ΔH (Nto0 (g)) = 2 * 33,2-81,6 = -15,2 kJ / mol
- Andre trinn: reaksjonsentropi-variasjonen
ΔSr = 2 * S (NOto (g)) - S (Nto0 (g)) - (3/2) S (Oto (g)) = 2 * 240,1 - 220,1 - 1,5 * 205,2 = -47,7 J / (mol * K).
- Tredje trinn: variasjon i Gibbs-funksjonen
Denne verdien vil bestemme balansen mellom avtagende energi og økende entropi for å vite om reaksjonen endelig er spontan eller ikke.
ΔGr = ΔHr - T ΔSr = -15,2 -298 * (- 47,7) = -985,4 J / mol
Siden det er en negativ variasjon av Gibbs-energien, kan det konkluderes med at det er en spontan reaksjon ved temperaturen 298 K = 25 ºC.


Ingen har kommentert denne artikkelen ennå.