De Canavan sykdom Det er en sjelden genetisk sykdom som oppstår fordi nerveceller i hjernen er skadet og ikke i stand til å kommunisere med hverandre. Denne sykdommen er til stede i ethvert samfunn og etnisk gruppe, selv om den er mye hyppigere i den jødiske befolkningen i Ashkenazi og deres etterkommere, hvor 1 av 6.400-13.000 mennesker er rammet. Den globale prevalensen er ukjent.
Denne sykdommen er innenfor gruppen leukodystrofier. Denne kategorien inkluderer alle genetiske forstyrrelser der myelinskjeden som omgir neuronaksonene er skadet, og det er derfor ingen god kommunikasjon mellom nevroner.
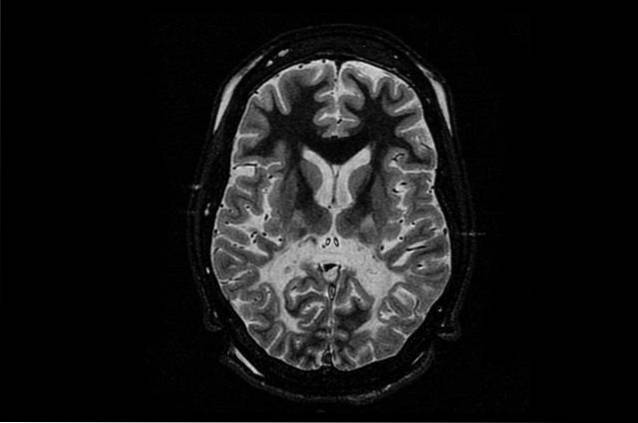
Den vanligste og samtidig mest alvorlige formen for denne sykdommen er nyfødt eller spedbarn. Denne formen for Canavan sykdom rammer nyfødte barn eller i deres første leveår..
Barn som lider av denne sykdommen gir ingen problemer i løpet av de første månedene av livet, men disse begynner å blomstre mellom 3 og 5 måneder. De viktigste symptomene skyldes utviklingsunderskuddet, der barn har motoriske problemer som hindrer dem i å snu seg, snu hodet eller sitte uten støtte..
Andre vanlige symptomer er muskelsvakhet (hypotoni), unormal hodeutvikling (makrocephaly) og irritabilitet. I mindre grad kan de også ha problemer med å spise, kramper og søvnproblemer..
En annen mindre vanlig form er Canavan sykdom som begynner i middelbarndommen eller ungdomsårene. Barn og ungdom med denne sykdommen har problemer med språkutvikling og motoriske ferdigheter, men disse problemene er ofte så milde at de ikke blir identifisert som symptomer på Canavan sykdom..
Forventet levealder for mennesker med Canavan sykdom er veldig heterogen, og varierer spesielt i henhold til tidspunktet for sykdommens begynnelse..
Barn som lider av nyfødt eller infantil form lever vanligvis bare noen få år, selv om noen når ungdomsårene og svært få til voksen alder. Mens de som lider av ungdomsformen har en normal forventet levetid.
Artikkelindeks
Det er to godt differensierte former for Canavan sykdom: den av nyfødt eller infantil debut og den av begynnelsen i middelbarndommen eller ungdomsårene..
Symptomer på nyfødt eller Canavan sykdom hos barn er svært alvorlige, vanligvis ikke merkbare før 3-50 måneder, og inkluderer makrocephaly, tap av motorisk kontroll av hodet og utviklingsmangel. Utviklingsunderskudd blir tydeligere etter hvert som barnet blir eldre.
De alvorligste symptomene er de som er relatert til motoriske problemer, siden barn ikke klarer å sitte eller stå opp uten støtte, gå eller snakke. Når de blir eldre kan hypotoni føre til spastisitet.
Selv om de har alle disse motoriske problemene, kan de lære å samhandle sosialt, smile, peke på gjenstander ...
Noen barn lider også av optisk atrofi, noe som forårsaker synsproblemer, selv om de fremdeles kan identifisere gjenstander visuelt.
Når symptomene vokser, blir de verre og forårsaker søvnvansker, kramper og problemer med å mate. Barnet blir helt avhengig og trenger hjelp til å utføre en hvilken som helst oppgave.
Forventet levealder for disse barna er ganske kort, de fleste dør på få år, selv om noen lever til ungdomsår eller voksen alder.
Canavan sykdom med begynnelse i middelbarndom eller ungdomsår er mildere enn den forrige. Symptomene inkluderer noen vanskeligheter i verbal og motorisk utvikling.
Selv om de vanligvis er så milde at de ikke blir identifisert som symptomer på Canavan sykdom, blir denne sykdommen vanligvis diagnostisert etter å ha utført en urinalyse, siden en av markørene er den høye konsentrasjonen av N-acetyl asparaginsyre (NAA i urinen)..
Denne sykdommen er forårsaket av en mutasjon i et gen kalt ASPA. Dette genet er det som kontrollerer enzymet aspartoacylase, som er ansvarlig for nedbrytende NAA-molekyler..
Mutasjonen av ASPA-genet fører til at aspartoacylase reduserer effektiviteten, så det vil ikke nedbryte nok NAA-molekyler, og det vil være en høy konsentrasjon av dette stoffet. Jo tidligere denne mutasjonen oppstår, jo verre effekter har den.
Selv om funksjonen til NAA-molekyler ikke er veldig godt forstått, ser det ut til at de er involvert i transport av vannmolekyler gjennom nevroner, og overskuddet av dette stoffet forhindrer at det dannes nytt myelin og ødelegger det eksisterende. Dette fører til at forbindelsene mellom nevroner ikke fungerer som de skal, og at hjernen ikke klarer å utvikle seg normalt..
Videre kan denne sykdommen arves på en autosomal recessiv måte. Så hvis hvert medlem av paret er bærer av den patogene varianten av ASPA-genet, og de bestemmer seg for å få et barn, vil de sannsynligvis:
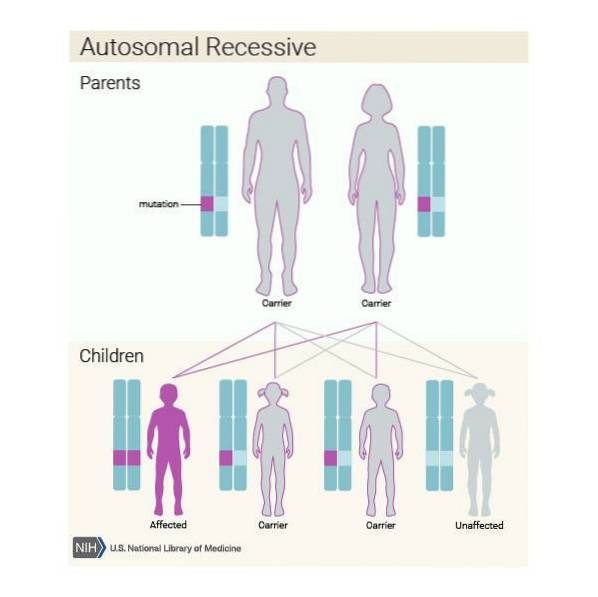
Det er veldig viktig at individer som tilhører risikopopulasjonen, i dette tilfellet etterkommere av Ashkenazi-jøder, gjennomgår en genetisk analyse for å sjekke om de bærer ASPA-genet før de får barn..
Behandlingen avhenger av sykdomsformen og symptomene hver enkelt presenterer..
Det er for tiden ingen kur mot Canavan sykdom, så tilgjengelige terapier fokuserer på å forbedre pasientens livskvalitet ved å støtte, pleie og hydrere, og forebygge og behandle infeksjoner.
Det anbefales at barn får fysioterapeutisk behandling for å forbedre kroppsholdning og motoriske ferdigheter, for å unngå og behandle kontrakturer og muskelproblemer, for eksempel trykksår. De kan også delta i terapeutiske og pedagogiske programmer for å forbedre sine kommunikasjonsevner..
Behandling med medisiner inkluderer antiepileptika (AED) hvis barnet har anfall, acetazolamid (merkenavn Diamox®) for å redusere intrakranielt trykk og injeksjoner av botulinumtoksin (Botox®) for å behandle spastisitet hvis den er tilstede.
Det er nødvendig å gjennomføre en oppfølging hver 6. måned for å sjekke hvilken tilstand barnet er i og hvordan utviklingen går.
Mennesker som lider av denne sykdomsformen, opplever mye mildere symptomer, så de trenger vanligvis bare terapier for å forbedre språket eller spesialpedagogiske programmer. De trenger ingen medisiner.
Årlig overvåking av barnets tilstand anbefales.
Effekten av andre terapier blir for tiden studert i både mennesker og dyremodeller..
Effekten av en genetisk transplantasjon til hjernen til barn med Canavan sykdom blir undersøkt ved hjelp av en ikke-viral vektor.
De første resultatene viser at denne typen transplantasjon tolereres godt av barn og forårsaker noen biokjemiske, radiologiske og metabolske forandringer, men det er ikke nyttig for å kurere sykdommen, så det utføres fortsatt tester (Leone et al 2000, Janson et al. . til 2002).
McPhee et al. (2006) gjennomfører en studie der det sunne ASPA-genet transplanteres til forskjellige steder i kropp av barn, med AAV2 som en vektor. I en av testene der 10 frivillige barn deltok. Hos 3 av dem virket transplantasjonen og nøytraliserte antistoffene, men ingen av barna ble bedre.
Litiumcitrat kan redusere nivået av NAA-konsentrasjon i hjernen, og det er derfor Assadi et al. (2010) bestemte seg for å gjennomføre et eksperiment der de administrerte litiumcitrat til 6 personer med Canavan sykdom i 60 dager.
NAA-konsentrasjonsnivåer i basalganglier og hvit substans i frontallappen ble funnet, selv om ingen kliniske forbedringer ble funnet.
Mangelen på aspartoacylaseenzymer forårsaker lave nivåer av acetat i hjernen, så Mahavarao og teamet hans (2009) bestemte seg for å gi glyseroltriacetat til to pasienter med Canavals sykdom for å øke acetatnivået og se om det også økte aspartoacylase-nivåene.
Forbindelsen ble godt tolerert av pasientene, selv om ingen kliniske forbedringer ble funnet. De gjennomfører for tiden tester som administrerer en høyere mengde glyseroltriacetat.
En av måtene å lage dyremodeller som representerer en sykdom, er å skape dyr slå ut. Disse dyrene, vanligvis mus, er genetisk modifisert for å fjerne eller endre genet som er endret i sykdommen. I dette tilfellet er det modifiserte genet ASPA-genet..
Dyremodeller brukes til å bedre forstå sykdommen, studere den biologiske korrelasjonen og verifisere effekten av nye behandlinger.
Matalon et al. (2003) brukte mus slå ut for å teste effekten av genterapi med AAV2 som vektor. De fant at det hadde vært forbedringer i myelinskjedene, men bare i noen deler, ikke i hele hjernen.
Surendrans team i samarbeid med Genzyme Corporation (2004) testet en stamcelletransplantasjonsbehandling. De fant at nye oligodendrocytter hadde blitt produsert, men ikke nok til å gjenopprette alle myelinskjedene..
Et annet team testet en terapi som besto av å erstatte de fungerende asparthoacyclase-enzymer med nye som ble injisert i bukhinnen på musene. slå ut.
De kortsiktige resultatene viste at enzymene klarte å passere blod-hjernebarrieren (når målet sitt) og var i stand til å redusere nivåene av NAA i hjernen betydelig. Selv om disse resultatene er lovende, er en langsgående studie nødvendig for å verifisere langtidseffektene (Zano et al., 2011).
De første tegnene som varsler leger om at noe er galt, er de fysiske, spesielt hypotoni og makrocephali.
Normalt, hvis disse tegnene blir observert, utføres en nevroimaging-studie vanligvis hos barnet for å sjekke for tegn på leukodystrofi, for eksempel en lavere tetthet av hvitt stoff. Det skal bemerkes at denne testen er mindre effektiv hos barn med Canavan sykdom som begynner i middelbarndommen eller ungdomsårene..
Når det er bevist at barnet lider av en leukodystrofi, gjøres mer spesifikke tester for å utelukke andre sykdommer, disse inkluderer:
Det siste trinnet for å bekrefte sykdommen ville være å utføre en genetisk studie som følger:


Ingen har kommentert denne artikkelen ennå.