
De Apert syndrom eller acrocephalosyndactyly type I (ACS1) er en patologi av genetisk opprinnelse som er preget av tilstedeværelsen av forskjellige endringer og misdannelser i hodeskallen, ansiktet og ekstremiteter.
På et klinisk nivå er Apert syndrom preget av tilstedeværelse eller utvikling av en spiss eller langstrakt hodeskalle, senket ansiktsområde med en endring i projeksjonen av tennene, fusjon og lukking av fingerben og ledd, mental retardasjon variabel, språkforstyrrelser , etc..
Selv om denne patologien kan være arvelig, oppstår Apert syndrom i de fleste tilfeller uten tilstedeværelse av familiehistorie, hovedsakelig på grunn av en de novo-mutasjon i svangerskapsfasen..
De genetiske mekanismene som forårsaker Apert syndrom er ikke akkurat kjent. Foreløpig er forskjellige genetiske endringer som er i stand til å produsere denne patologien identifisert, hovedsakelig relatert til mutasjoner i FGFR2-genet.
På den annen side begynner diagnosen Apert syndrom vanligvis med klinisk mistanke i prenatalperioden etter identifisering av abnormiteter i rutinemessige ultralydskanninger, og bekreftes ved å utføre en genetisk studie.
Når det gjelder behandling, er det ingen form for kurativ intervensjon for Apert syndrom. Imidlertid har det gjennom historien til denne patologien blitt utviklet forskjellige spesifikke inngrep som vanligvis inkluderer nevrokirurgi, kraniofacialkirurgi, kjeve-, ansiktskirurgi, medikamentell behandling, fysioterapi, psykologisk og nevropsykologisk intervensjon, blant andre.
Artikkelindeks
Apert syndrom er en genetisk patologi preget av tilstedeværelsen av forskjellige skjelettmisdannelser på kranial-, ansikts- og / eller lemmerivå.
Den vesentlige endringen av Apert syndrom består av en tidlig eller tidlig lukking av kraniale sprekker, noe som forårsaker en unormal vekst av resten av strukturene i ansiktet og hodeskallen. I tillegg til disse kan det oppstå misdannelser i øvre og nedre ekstremiteter, for eksempel sammensmelting av fingre og tær..
På den annen side kan de kognitive evnene til personer som lider av Apert syndrom også påvirkes, med variabel alvorlighetsgrad fra mild til moderat.
Selv om Baumgartner (1842) og Wheaton (1894) nevnte den første om denne medisinske tilstanden, var det først i 1906, da den franske medisinske spesialisten Eugene Apert, nøyaktig beskrev dette syndromet og publiserte den første kliniske rapporten..
I sin publikasjon, Eugene Apert, beskriver han et sett med nye tilfeller av pasienter som er rammet av et veldefinert misdannelsesmønster og preget av de karakteristiske tegn og symptomer på denne patologien..
Dermed var det først i 1995 at de etiologiske genetiske faktorene til Apert syndrom ble identifisert. Spesielt beskrev Wilkie et al. Tilstedeværelsen av to mutasjoner i FGFR2-genet hos rundt 40 berørte pasienter..
I tillegg er Apert syndrom en medisinsk tilstand som er klassifisert i sykdommene eller patologiene som er preget av å presentere kraniosynostose (for tidlig lukking av kraniale suturer).
Andre patologier som tilhører denne gruppen er Pfeiffer syndrom, Crouzon syndrom, Saethre-Chotzcen syndrom og Carpenter syndrom..
Apert syndrom regnes som en sjelden eller sjelden patologi, det vil si at den har en forekomst på mindre enn ett tilfelle per 15.000 innbyggere i befolkningen generelt..
Spesielt forekommer Apert syndrom rundt en person for hver 160.000-200.000 fødsler, og i tillegg er det 50% sannsynlighet for å overføre denne patologien på arvelig nivå.
I tillegg, med hensyn til fordeling etter kjønn, er det ikke identifisert en høyere forekomst hos menn eller kvinner, og den har heller ikke vært assosiert med bestemte etniske grupper eller geografiske steder..
For tiden, og gitt at Apert syndrom ble identifisert i omtrent 1984, i kliniske rapporter og i medisinsk litteratur som har publisert mer enn 300 tilfeller av denne patologien..
De kliniske manifestasjonene av Apert syndrom inkluderer vanligvis en misdannelse eller ufullstendig utvikling av kraniestrukturen, en atypisk fenotype eller ansiktsmønster og skjelettendringer i ekstremiteter..
Når det gjelder Apert syndrom, er den sentrale involveringen relatert til dannelsen og lukkingen av benets struktur i hodeskallen. Under embryonal utvikling forekommer en prosess kalt creneosynostosis, preget av for tidlig lukking av kraniale suturer.
Kranifissurer eller suturer er en type fibrøse vevsbånd som har det grunnleggende målet å forbinde bein som utgjør hodeskallen (frontal, occipital, parietal og temporal).
I løpet av svangerskapsfasen og den tidlige postnatale perioden holdes benstrukturen som utgjør hodeskallen sammen takket være disse fibrøse og elastiske vevene.
Normalt smelter kranialbenet ikke før rundt 12 til 18 måneder. Tilstedeværelsen av myke flekker eller mellomrom mellom hodebenene er en del av normal utvikling av barn.
I løpet av hele barndomsfasen lar disse suturene eller de fleksible områdene hjernen vokse på en akselerert måte og i tillegg beskytter den mot støt.
I Apert syndrom gjør for tidlig lukking av disse kraniale suturene og kranialbenet normal utvikling av hjerne- og hjernevekst umulig..
Følgelig kan de vanligste tegnene og symptomene på Apert syndrom omfatte:
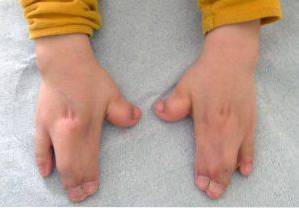
Disse typer endringer påvirker hovedsakelig øvre og nedre ekstremiteter, vanligvis fusjon og utvikling av fingrene..
I tillegg til disse er det også mulig å observere andre kliniske funn på muskuloskeletalt nivå, forkortelse av forskjellige bein (radius, humerus, lårben), hypoplasi i scapula eller bekken, fusjon av livmorhvirvler.
Som en konsekvens vil mange berørte ha redusert leddmobilitet og kan derfor utvikle ulike vanskeligheter for å tilegne seg grov- og finmotorikk..
Disse typer avvik er veldig heterogene og varierer blant berørte individer, men noen av de vanligste har blitt identifisert:
Den etiologiske endringen av denne patologien kan føre til utvikling av lesjoner eller sekundære patologier på et morfologisk og strukturelt nivå i forskjellige områder av kroppen, noen av dem inkluderer:
Til tross for at det i mange tilfeller er mulig å observere tilstedeværelsen av en generell endring av kognitive funksjoner og intellektuelt nivå, er mental retardasjon ikke utvetydig til stede i alle tilfeller av Apert syndrom..
I tillegg, i tilfeller der det er en forringelse av det intellektuelle nivået, kan dette være variabelt, på en skala fra mild til moderat.
På det språklige området er utviklingen av ulike underskudd ofte, hovedsakelig relatert til artikulering av lyder som et resultat av mandibulære og orale misdannelser..
Apert syndrom skyldes tilstedeværelsen av en spesifikk mutasjon i FGFR2 genet. Eksperimentelle studier har indikert at dette genet er ansvarlig for produksjonen av et protein, kalt fibroblast vekstfaktorreseptor 2.
Blant funksjonene til denne faktoren beskriver den sending av forskjellige kjemiske signaler til umodne celler for å forårsake transformasjon og differensiering i beinceller under fosterets eller prenatal fase av utviklingen..
Derfor forandrer tilstedeværelsen av mutasjoner i FGFR2-genet funksjonen til dette proteinet, og kan derfor forårsake tidlig fusjon av bein i hodeskallen, hånden og føttene..
En god del av de kliniske egenskapene til Apert syndrom kan identifiseres under graviditet, spesielt ved ultralydundersøkelser av graviditet og fosterutvikling..
Når det er klinisk mistanke, startes en genetisk studie på nytt for å identifisere tilstedeværelsen av en genetisk mutasjon som er kompatibel med Apert syndrom..
På den annen side, når tegnene er subtile eller ikke har blitt identifisert før fødselen, er det etter dette mulig å utføre en detaljert fysisk analyse og forskjellige genetiske tester for å bekrefte diagnosen..
Selv om det ikke er noen spesifikk kur mot Apert syndrom, har forskjellige tilnærminger blitt beskrevet for behandling av symptomer og medisinske komplikasjoner ved denne patologien..
De mest effektive terapeutiske inngrepene er de som implementeres tidlig, i de første øyeblikkene av livet og som involverer fagpersoner fra forskjellige områder.
Vanligvis krever behandling av berørte barn individuell planlegging, med flere operasjoner planlagt. Dermed er håndteringen av denne patologien basert på korreksjon av skjelett- og kranio-ansikts misdannelser, og psykologisk og nevropsykologisk støtte..
Gjennom nevrokirurgi er målet å rekonstruere kranialhvelvet, mens spesialister innen kjeve- og ansiktskirurgi prøver å korrigere misdannelser i ansiktet. På den annen side er traumekirurger ofte med, for rekonstruksjon av misdannelser i hender og føtter.
I tillegg er utformingen av individualiserte programmer for tidlig stimulering, kommunikasjonsrehabilitering, sosial ferdighetstrening eller psyko-pedagogisk oppfølging gunstig for å oppnå en optimal, funksjonell og uavhengig utvikling av de berørte individene..



Ingen har kommentert denne artikkelen ennå.