
De Prader-Willi syndrom (SPW) er en multisystemisk patologi som har medfødt genetisk opprinnelse. Det er en kompleks sykdom som påvirker appetitt, vekst, metabolisme, atferd og / eller kognitiv funksjon.
På klinisk nivå, i barndomsfasen, er denne sykdommen preget av tilstedeværelsen av forskjellige medisinske funn som muskelsvakhet, spiseforstyrrelser eller generalisert utviklingsforsinkelse..
I tillegg presenterer en god del av individene som er rammet av Prader-Willi syndrom på kognitivt og atferdsmessig nivå en moderat intellektuell svekkelse eller forsinkelse som er ledsaget av ulike lærings- og atferdsproblemer..
Selv om Prader-Willi syndrom regnes som en sjelden eller uvanlig sykdom, indikerer mange studier at det er en av de hyppigste patologiene i det genetiske området. Diagnosen av denne sykdommen stilles hovedsakelig på grunnlag av kliniske funn og komplementære genetiske tester..
Når det gjelder behandling, er en kur mot Prader-Willi syndrom ennå ikke identifisert, så den terapeutiske tilnærmingen er orientert mot behandling av symptomer og komplikasjoner, med fedme som det medisinske funnet som utgjør den største trusselen for de berørte.
I forhold til prognose og livskvalitet, vil begge avhenge av alvorlighetsgraden av de tilknyttede medisinske problemene og de atferds- eller kognitive lidelser som kan utvikle seg..
Artikkelindeks
Ulike kliniske rapporter indikerer at Prader-Willi syndrom (PWS) opprinnelig ble beskrevet av J. L. Down, i 1887, etter å ha diagnostisert en av pasientene med “polysarcia”..
Imidlertid var det Drs Prader, Labhart og Willi som i 1956 beskrev ytterligere 9 tilfeller og ga denne patologien navnet sitt. I tillegg ble egenskapene og diagnostiske kriteriene for Prader-Willi syndrom systematisert av Holm et al..
Prader-Willi syndrom er en medfødt genetisk endring, det vil si at det er en patologi som er tilstede fra fødselsøyeblikket og vil påvirke individet gjennom hele livet hvis det ikke er noen kurativ terapeutisk inngrep..
Denne patologien presenterer et komplekst klinisk forløp, preget av mange medisinske manifestasjoner.
Selv om fenotypen til Prader-Willi syndrom i dag er mer kjent, har det vært de siste 25 årene, da det har vært betydelige fremskritt i analysen og forståelsen av denne sykdommen..
Uttrykket av Prader-Willis syndrom er mangfoldig, det har en tendens til å påvirke flere systemer og strukturer, de fleste av endringene er relatert til hypotalamus dysfunksjon.
Hypothalamus er en nevrologisk struktur som har en viktig rolle i kontrollen av homeostatiske funksjoner: regulering av sult, tørst, søvn-våknesyklus eller regulering av kroppstemperatur.
I tillegg frigjør hypothalamus forskjellige hormoner til forskjellige kjertler: vekst, seksuell, skjoldbruskkjertel, etc..
Til slutt må vi påpeke at Prader-Willis syndrom også kan vises referert i medisinsk og eksperimentell litteratur med andre begreper som Prader-Labhart-Willi syndrom eller med akronymet PWS..
Også andre synonymer er Labhart Willi syndrom, Praser Labhart Willi Fancone syndrom eller hypogenital dystrofi syndrom..
Prader-Willi syndrom (PWS) er en sjelden genetisk sykdom. Uttrykket sjelden sykdom (ER) brukes til å referere til de patologiene som er sjeldne eller få mennesker som lider av den.
Foreløpig anslås det at Prader-Willi syndrom er en patologi med en omtrentlig frekvens på 1 tilfelle per 10.000-30.000 mennesker over hele verden.
På den annen side, når det gjelder fordeling etter kjønn, har det blitt observert at denne patologien påvirker menn og kvinner likt, og den er ikke assosiert med etniske grupper eller geografiske regioner..
I tillegg betraktes Prader-Willi syndrom som den viktigste årsaken til fedme av genetisk opprinnelse.
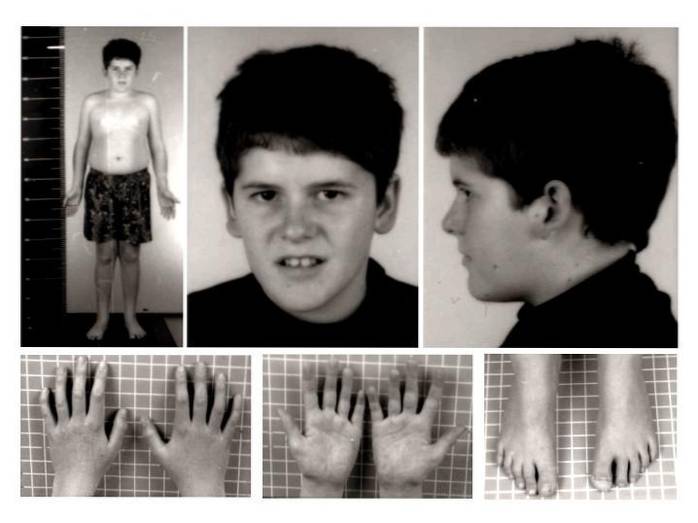
På klinisk nivå har Prader-Willi syndrom tradisjonelt vært assosiert med nyfødt hypotoni, hypogonadisme, hyperfagi, fedme, kort vekst, generalisert forsinkelse i utviklingen, moderat utviklingshemning, atypisk ansiktsutseende og forskjellige atferdsendringer..
Til tross for dette er det kliniske uttrykket for denne patologien veldig heterogen og varierer betydelig mellom berørte individer..
I tillegg har de karakteristiske tegn og symptomer på Prader-Willi syndrom en tendens til å variere med biologisk utvikling, slik at vi kan observere forskjellige kliniske funn i foster- og nyfødtperioden, spedbarnsperioden eller tidlig barndom, skolestadiet og til slutt scenen Teen.
På en systematisk måte beskriver José A. del Barrio del Campo og samarbeidspartnere i detalj de mest karakteristiske endringene i det biomedisinske, psykomotoriske, kognitive og atferdsmessige området:
De mest karakteristiske fysiske tegn og symptomer inkluderer forstyrrelser som; hypotoni, misdannelser i muskler og skjelett, redusert eller lav vekt og høyde, overflødig appetitt, fedme, hypogonadisme, søvnforstyrrelser, luftveissykdommer, atypiske lette egenskaper, endring i regulering av kroppstemperatur, blant andre.
Tilstedeværelse eller utvikling av redusert muskeltonus. Den muskuløse slappheten i denne patologien er spesielt fremhevet i nakken og kofferten, spesielt i nyfødtstadiet og de første månedene av livet. Dermed, med biologisk utvikling, har muskeltonen en tendens til å bli bedre.
I dette tilfellet er det vanlig å observere utviklingen av skoliose eller avvik i ryggraden, dårlig justering av underekstremiteter (genu valgus) eller tilstedeværelsen av flate føtter..
I tillegg kan andre typer medfødte anomalier også observeres, som reduksjon i størrelse på føtter og hender, hoftedysplasi, tilstedeværelse av seks fingre, blant andre..
Spesielt på fødselstidspunktet er både høyden og vekten til det berørte barnet lavere enn forventet for deres utvikling og kjønn. Til tross for at standardverdier kan nås i voksen alder, har den langsomme vekstraten en tendens til å endre voksenverdier for høyde og vekt.
Det er vanlig å observere en umettelig appetitt hos personer med Prader-Willi syndrom, preget av en besettelse eller fiksering på maten. På grunn av inntak av store mengder mat, har de som er rammet en tendens til å utvikle fedme og andre tilknyttede medisinske komplikasjoner, som type II diabetes mellitus.
Tilstedeværelsen av kjønnsendringer er også hyppig. Spesielt er hypogonadisme eller delvis utvikling av de ytre kjønnsorganene veldig vanlig. I de fleste tilfeller klarer ikke pubertetsutviklingen å nå det endelige eller voksenstadiet.
Snorking, økt frekvens eller åndedrettsstopp vises ofte i søvnfaser. Dermed har de berørte en tendens til å presentere forskjellige endringer relatert til fragmentering, søvnforsinkelse eller tilstedeværelsen av periodiske oppvåkning.
Muskuloskeletale abnormiteter og misdannelser kan også påvirke kraniofaciale funksjoner. Det er mulig å observere en smal hodeskalle, øye-strabismus, dårlig pigmentert hud og hår, liten munn og tynne lepper, tannskader etc..
Personer som er rammet av Prader-Willi syndrom har vanligvis problemer relatert til regulering av kroppstemperatur, og et annet viktig funn er høy motstand mot smerte..
På grunn av tilstedeværelsen av muskel- og skjelettdeformasjoner og redusert muskeltonus, vil psykomotorisk utvikling gå langsommere, og påvirke alle områder.
De berørte presenterer vanligvis serievansker med å utføre alle typer aktiviteter som krever en eller flere motoriske henrettelser.
Når det gjelder kognitive begrensninger, har de fleste av dem som er rammet en mild eller moderat utviklingshemming.
I tillegg til dette presenterer de vanligvis noen spesifikke områder som er mer berørt, slik som sekvensiell prosessering av informasjon, nylig eller korttidsminne, løsning av aritmetiske problemer, auditiv prosessering av verbal informasjon, endring av oppmerksomhet og konsentrasjon og tilstedeværelse av kognitiv stivhet.
På den annen side er språk et annet område som påvirkes betydelig hos personer med Prader-Willi syndrom. Forsinkelser i tilegnelsen av fonologiske ferdigheter, dårlig ordforråd, endring av blant annet grammatisk konstruksjon blir vanligvis observert..
Atferdsproblemer og endringer er et annet av de typiske funnene som kan observeres i Prader-Willi syndrom, de varierer normalt avhengig av alder eller modningsstadium der den berørte personen er, men noen av de vanligste atferdstrekkene er:
Ulike nåværende undersøkelser har påpekt at atferdsendring har en tendens til å øke med alderen, og derfor har de en tendens til å forverres, og påvirker sosiale, familie- og følelsesmessige områder på en generalisert måte.
Som vi har påpekt i flere seksjoner ovenfor, har Prader-Willi syndrom en genetisk opprinnelse.
Selv om det for tiden er en stor kontrovers om de spesifikke gener som er ansvarlige for denne patologien, viser alle data at den etiologiske endringen er lokalisert på kromosom 15..
Gjennom den genetiske studien av denne patologien har det vært flere bidrag. Burtler og Palmer (1838) oppdaget tilstedeværelsen av abnormiteter i den lange armen av kromosom 15 fra farens foreldre, mens Nicholls (1989) observerte at lidelsen i andre tilfeller var relatert til kromosomale endringer fra moren (Rosell-Raga, 2003).
Bortsett fra dette, er den mest aksepterte teorien om opprinnelsen til denne patologien tap eller inaktivering av forskjellige gener av faderlig uttrykk som er lokalisert i kromosom 15-regionen 15q11-13..
Diagnosen av Prader-Willi syndrom har to grunnleggende komponenter, analyse av kliniske funn og genetisk testing..
Når det gjelder påvisning av indikatortegn og symptomer, både hos babyer og hos eldre barn, vil det være viktig å utføre en detaljert, individuell og familiemedisinsk historie. På samme måte er det også viktig å utføre en fysisk og nevrologisk undersøkelse..
Hvis det er en diagnostisk mistanke, basert på disse prosedyrene, vil det være nødvendig å foreskrive forskjellige komplementære tester for å bestemme tilstedeværelsen av genetiske endringer og abnormiteter..
Nærmere bestemt blir rundt 90% av tilfellene definitivt diagnostisert gjennom DNA-metyleringstester og andre tilleggstester..
I tillegg er det også mulig å stille en prenatal diagnose av denne medisinske tilstanden, hovedsakelig i familier med en tidligere historie med Prader-Willi syndrom..
Nærmere bestemt tillater fostervannsprøven ekstraksjon av embryoprøver for utførelse av relevante genetiske tester.
Det er for øyeblikket ingen kur mot Prader-Willi syndrom. Som i andre sjeldne sykdommer, er behandlinger begrenset til symptomkontroll og forbedring av livskvaliteten til berørte mennesker.
Imidlertid vil en av de grunnleggende aspektene være ernærings- og diettkontroll, siden fedme er den viktigste årsaken til sykelighet og dødelighet i denne patologien..
På den annen side vil tilstedeværelsen av kognitive og atferdsmessige endringer kreve intervensjon av spesialiserte fagpersoner både i kognitiv rehabilitering og i håndtering av atferdsforstyrrelse.



Ingen har kommentert denne artikkelen ennå.