De Cornelia de Lange syndrom er en patologi av genetisk opprinnelse som er preget av tilstedeværelsen av en betydelig kognitiv forsinkelse ledsaget av forskjellige misdannende fysiske egenskaper.
På klinisk nivå observeres tre forskjellige kliniske forløp: alvorlig, moderat og mild. Tegn og symptomer består vanligvis av en atypisk ansiktskonfigurasjon, muskuloskeletale misdannelser og forsinket kognitiv og psykomotorisk utvikling. I tillegg kan andre typer abnormiteter relatert til hjerte-, lunge- og / eller fordøyelsesdeformasjoner skille seg ut..
Når det gjelder opprinnelsen til Cornelia de Lange syndrom, har dets etiologi vært relatert til tilstedeværelsen av spesifikke mutasjoner i genene SMC3, SMC1A, NIPBL, blant andre. Diagnosen er grunnleggende klinisk, basert på fysiske og kognitive egenskaper. Imidlertid er det vanligvis ledsaget av en bekreftende genetisk test.
Behandlingen er rettet mot påvisning og behandling av medisinske komplikasjoner. Medisin, logoped, nevropsykologisk inngrep og spesialundervisning er avgjørende.
Artikkelindeks
Dette syndromet ble opprinnelig beskrevet av Dr. Cornelia de Lange i 1933. Hennes forskning var basert på studien av to pasienter i alderen 6 og 17 måneder. Hans kliniske bilde var preget av en alvorlig forsinkelse i fysisk vekst og intellektuell utvikling assosiert med forskjellige misdannende egenskaper..
Gitt likheten i begge tilfeller antok den første kliniske rapporten om denne patologien eksistensen av en felles og offentlig etiologisk årsak.
Tidligere hadde Brachmann (1916) klart å publisere noen obduksjonsdata for en pasient i barndommen med noen egenskaper som er kompatible med Cornelia de Lange-syndromet..
For tiden er det kliniske bildet av dette syndromet klassifisert i tre differensielle fenotyper: alvorlig, moderat og mild..
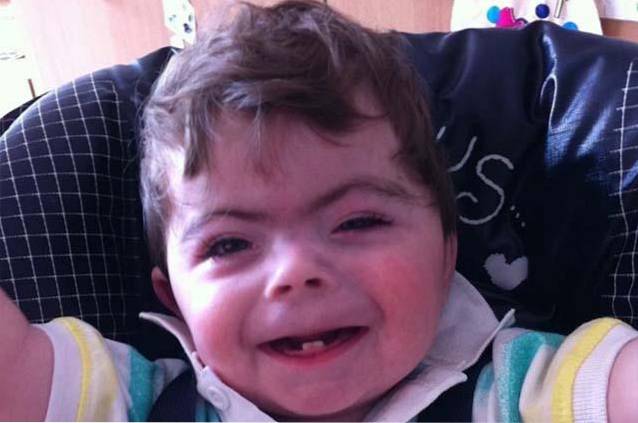
Cornelia de Lange syndrom er en sjelden genetisk lidelse av medfødt karakter, det vil si at dens kliniske egenskaper er tydelige fra fødselen. Det er definert som en multisystemisk sykdom med symptomer assosiert med forsinket fysisk og kognitiv utvikling, kranio-ansikts misdannelser eller muskuloskeletale misdannelser..
Selv om det kliniske forløpet og alvorlighetsgraden av dette syndromet kan variere betydelig blant de berørte, er det en sykdom med høy dødelighet.
Personer med Cornelia de Lange syndrom er preget av å ha en atypisk eller karakteristisk ansiktskonfigurasjon og en forsinkelse i vekst / utvikling før og etter fødselen..
Det vanligste er at læringsproblemer dukker opp, forsinket språkoppkjøp eller gangart og atferdsmessige avvik.
Cornelia de Lange syndrom er en sjelden sykdom i den generelle befolkningen, den er vanligvis klassifisert i sjeldne sykdommer. Epidemiologiske data er ikke akkurat kjent. Forekomsten er estimert til ett tilfelle per 10.000-30.000 fødsler..
Til dags dato kan vi finne mer enn 400 forskjellige tilfeller av Cornelia de Lange syndrom beskrevet i medisinsk og eksperimentell litteratur..
Det er en patologi som kan påvirke begge kjønn i like mange. Noen forfattere som Gutiérrez Fernández og Pacheco Cumani (2016) antyder en liten overvekt overfor kvinner, med et forhold på 1,3 / 1.
I forhold til resten av sosiodemografiske faktorer har dagens forskning ikke identifisert en differensiell prevalens assosiert med spesifikke land eller etniske og / eller rasegrupper..
En god del av de diagnostiserte tilfellene er sporadiske, selv om flere berørte familier har blitt identifisert med et tydelig mønster av dominerende arv..
Tegn og symptomer på Cornelia de Lange syndrom er preget av deres brede mønster av involvering.
Denne sykdommen er definert av tilstedeværelsen av karakteristiske ansiktsegenskaper, muskuloskeletale misdannelser i øvre og nedre lemmer, generalisert pre- og postnatal veksthemming, sammen med utvikling av andre fysiske abnormiteter..
Deretter vil vi beskrive noen av de hyppigste kliniske trekkene ved Cornelia de Lange syndrom:
Hos mer enn 90% av de som er rammet av Cornelia Lange syndrom, er det mulig å identifisere en forsinkelse i fysisk utvikling eller global hypogrowth. Vekst påvirkes vanligvis både prenatalt og postnatalt.
De vanligste egenskapene hos nyfødte er:
Disse forholdene varer vanligvis til voksen alder. I den kan man skille en vekst som ligger under den forventede for den berørte personens kjønn og biologiske alder.
Sammen med denne typen endringer kan det identifiseres noen abnormiteter knyttet til fôring. Vanskeligheter med å svelge eller tygge mat er vanlig i de tidlige stadiene av livet.
Kombinasjonen av kraniale og ansiktsendringer resulterer i utviklingen av en karakteristisk ansiktsfenotype hos personer som lider av Cornelia de Lange syndrom..
Noen av de vanligste abnormitetene inkluderer:
Forsinkelse i kognitiv og psykomotorisk utvikling er et av de sentrale kliniske funnene i Cornelia Lange syndrom. En langsom tilegnelse av ferdigheter knyttet til motorisk eller mental aktivitet blir vanligvis identifisert.
De mest berørte milepælene er anskaffelse av sittende, det affektive smilet, pludringen, den uavhengige bevegelsen, utslipp av de første ordene, forståelse og ordrer, fôring, ambulation eller uavhengig toalett.
Hos mange av de berørte kan en gjennomsnittlig IQ assosiert med mild eller moderat utviklingshemming identifiseres.
Oppførselen til de som er rammet av Cornelia de Lange syndrom presenterer vanligvis noen særegenheter:
Cornelia de Lange syndrom er også forbundet med utvikling av ulike medisinske komplikasjoner.
De hyppigste dødsårsakene eller forverringen av den medisinske statusen til de berørte er relatert til:
Variasjonen av tegn og symptomer på Cornelia de Lange syndrom har tillatt en klassifisering av dets kliniske forløp:
Det er vanligvis det mest alvorlige. Endringene og anomaliene er preget av tilstedeværelse av fysisk undervekst, misdannelser i skjelett, unormale ansiktsegenskaper, begrensning av leddets mobilitet, kognitiv forsinkelse og andre medisinske komplikasjoner (auditiv, okulær, fordøyelsessystem, reno-urologisk, hjerte- og kjønnsorgan)..
I denne undertypen er de fysiske endringene vanligvis ikke veldig tydelige, spesielt i ekstremiteter. De berørte har vanligvis ikke alvorlige intellektuelle underskudd. Den vanligste er at diagnosen stilles utover nyfødtstadiet.
Dens kliniske forløp er fundamentalt preget av klinisk variasjon. Ansiktsegenskaper er til stede i de fleste tilfeller, men uttrykket for resten av anomaliene er variabelt..
Opprinnelsen til Cornelia Lange syndrom er forbundet med tilstedeværelsen av genetiske abnormiteter. I de undersøkte tilfellene var det mulig å identifisere spesifikke mutasjoner i 5 forskjellige gener: NIPBL, SMC1A, HDAC8, RAD21 og SMC3.
Den vanligste endringen er relatert til NIPBL-genet, identifisert hos mer enn halvparten av de berørte. Resten av genetiske anomalier er sjeldnere.
Alle disse genene spiller en fremtredende rolle i produksjonen av proteiner relatert til kohesinkomplekset, ansvarlig for regulering av kromosomstruktur og organisering, stabilisering av genetisk informasjon i celler og DNA-reparasjon..
I tillegg oppfyller de også flere grunnleggende funksjoner i prenatal utvikling av ekstremiteter, ansikt og andre regioner og kroppssystemer.
Diagnosen Cornelia de Lange syndrom er klinisk. Foreløpig er det ingen laboratorietest som definitivt indikerer tilstedeværelsen. I det medisinske området er det vanligste å bruke diagnostiske kriterier foreslått av Kline et al..
Disse refererer til identifisering av kraniofaciale anomalier, i vekst og utvikling, i ekstremiteter, nevrosensoriske og kutane endringer, atferdsforstyrrelser, etc..
I tillegg er det viktig å utføre en molekylærgenetisk analyse for å identifisere tilstedeværelsen av mutasjoner assosiert med Cornelia de Lange syndrom..
Selv om det ikke finnes noen kur mot Cornelia de Lange-syndromet, innebærer dets terapeutiske tilnærming utforming av en kontinuerlig medisinsk oppfølging sammen med behandling av komplikasjoner..
Forfatterne Gil, Ribate og Ramos (2010) peker på noen av de mest brukte tilnærmingene.


Ingen har kommentert denne artikkelen ennå.